
OHIO researcher uses desert flowers to pursue an unanswered question about macroevolution
Environmental & Plant Biology Department
Students explore yesterday, today and tomorrow, from the fossil records of flora from the past to how plants react to lack of gravity in space. They study plants at a molecular and genetic level and look for solutions to environmental sustainability.
Undergraduate DegreesGraduate Degrees
With a Plant Biology Degree...
-
'Lichen' Your InternshipSurveying proposed mountain bike trails in Wayne National Forest and documenting invasive species.
-
Plant a Fertilizer?Studying peanut plants' potential as a natural nitrogen fertilizer, reducing fertilizer costs and agricultural runoff.
-
Save a Tree?Researching the genetic diversity of ash trees in search of a key to resisting the Emerald ash borer.
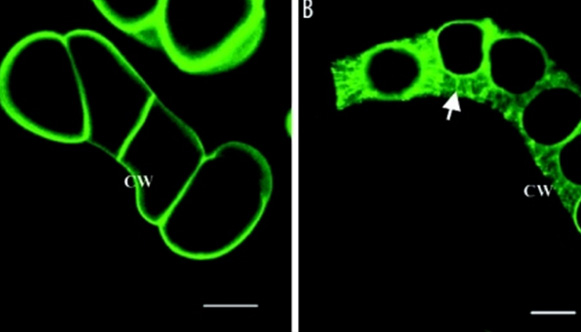
Functional Genomics
Using molecular, genetic and genomic tools to study plants.

Ecology
Using the ecosystems of southeastern Ohio and beyond as a natural laboratory.

Systematics and Evolution
Discovering new species, understanding evolution, finding biogeographic trends.

Undergraduate Research
Conducting undergraduate research on campus and beyond—plus internships.

Sustainability in Practice
Learning at the garden and in the greenhouse.

Community Activities
Engaging the public—especially children and high school students—in science.